Previously we had successfully run GWAS on almost all of the UKBiobank traits (https://ieup4.blogs.bristol.ac.uk/2018/10/01/ukb_gwas/). Our next job was to make these searchable at scale. This post explains how we have done this and how you can access the data.
Background
MR-Base (http://www.mrbase.org/) is a platform for performing 2-sample Mendelian Randomization to infer causal relationships between phenotypes. In its beginnings GWAS were manually curated and loaded into a database for use with the platform. Each GWAS (after QC) consisting of a set number of columns (9) and a variable number of rows (typically in the millions, for UKBiobank 10 million). This process quickly became problematic due to large numbers of GWAS and increasing numbers of users. For those interested in how we managed to successfully implement the indexing of over 20,000 GWAS, including each problem we encountered along the way, you can read the details of the journey below. For those who aren’t, you can skip to the end.
Moving forward
Initially we were working with 100s of GWAS, so MySQL was doing OK. As the data grew we tried to improve things using different table formats and the like, but it quickly became unsuitable. We tested a few different ideas, including Neo4j and HDF5 files but settled on Elasticsearch. As this was basically a simple lookup of large amounts of uniform data an index seemed the right way to go. We also needed to query in multiple ways, e.g. by SNP ID, by Study ID and by P-value which made HDF5 difficult to implement. An initial setup for Elasticsearch on a single server was quick and indexing a relatively small number of GWAS (~1,000) was quick.
Problem 1
The data were indexed but search speed was awful. As the Study ID was an integer it had been indexed in that format. This, however, was not good for searching, as numbers are treated differently:
The fact that some data is numeric does not mean it should always be mapped as a numeric field. The way that Elasticsearch indexes numbers optimizes for
rangequeries whilekeywordfields are better attermqueries. Typically, fields storing identifiers such as anISBNor any number identifying a record from another database are rarely used inrangequeries or aggregations. This is why they might benefit from being mapped askeywordrather than asintegerorlong.
So, once that was changed, search was quick.
Problem 2
Once we were happy with performance, and anticipated 1,000s more GWAS (https://ieup4.blogs.bristol.ac.uk/2018/10/01/ukb_gwas/) we decided to buy some more servers. We bought 4 standard machines, each with 384 GB RAM and 24/40 cores. Three were set up as master/data nodes and one as an interface node. The three data nodes were set up with RAID0 and data integrity was to be managed by Elasticsearch, e.g. if one node goes down, the data from that node are served by the other two machines. Copying the data over from the old server was relatively straight forward by taking a snapshot and restoring on the new servers. The new machines gave us some space and time to test different strategies for indexing the data.
The standard Elasticsearch variables are number of indexes, shards and replicas (https://www.elastic.co/guide/en/elasticsearch/reference/current/getting-started-concepts.html). We were planning to separate each data batch into different indexes but were not sure how best to split the data into shards/replicas. Lots of small shards permit lots of parallel compute, but the data from each queried shard needs to be compiled into a single data set. After trying a few large indexes with many shards, and lots of small ones with few shards, we decided to do some reading. While there is nothing definitive about how to setup a cluster, there are guidelines:
https://www.elastic.co/guide/en/elasticsearch/guide/current/multiple-indices.html
Searching 1 index of 50 shards is exactly equivalent to searching 50 indices with 1 shard each: both search requests hit 50 shards.
https://qbox.io/blog/optimizing-elasticsearch-how-many-shards-per-index
…one shard per 30 GB of data.
https://www.elastic.co/guide/en/elasticsearch/reference/current/getting-started-concepts.html
Each Elasticsearch shard is a Lucene index. There is a maximum number of documents you can have in a single Lucene index. As of
LUCENE-5843, the limit is2,147,483,519(= Integer.MAX_VALUE – 128) documents. You can monitor shard sizes using the_cat/shardsAPI.
https://www.elastic.co/blog/how-many-shards-should-i-have-in-my-elasticsearch-cluster
TIP: The number of shards you can hold on a node will be proportional to the amount of heap you have available, but there is no fixed limit enforced by Elasticsearch. A good rule-of-thumb is to ensure you keep the number of shards per node below 20 per GB heap it has configured. A node with a 30GB heap should therefore have a maximum of 600 shards, but the further below this limit you can keep it the better. This will generally help the cluster stay in good health.
This last tip was particularly interesting, and also worrying. It appeared we were bound by the fixed limit of heap, which can be no more that 32GB. In theory this limited us to a set number of shards, each of which had a recommended maximum size. For examples:
20 shards per GB of heap = 600 shards for 30 GB
x 3 for 3 nodes = 1,800 shards
Max GB disk per shard = 50 GB
1,800 X 50 GB = 90 TB
If 1 GB = 1 study, 90 TB = 9000 studies
As we were planning on indexing 10s of 1,000s of studies, this didn’t look good. The machines we were using were not ideal, Elasticsearch is designed to scale horizontally with more machines and we had 3 large machines. At this point, it would have been wise to figure out how to run multiple instances on each node, but instead, we carried on regardless, hoping that the recommended maximums were conservative. In addition, the data were now being queried quite heavily, and every time we tried something experimental we risked killing Elasticsearch and down time for those tools that were using it.
We settled on the 3 shards, each with one replica. Indexing the UKBiobank GWAS was going OK until things got very slow at about 10,000 studies and then stopped. We had hit limits with indexing speed and segment number (each shard is split into segments which requires a small amount of heap). This had created two new problems, indexing speed and heap issues.
Problem 3
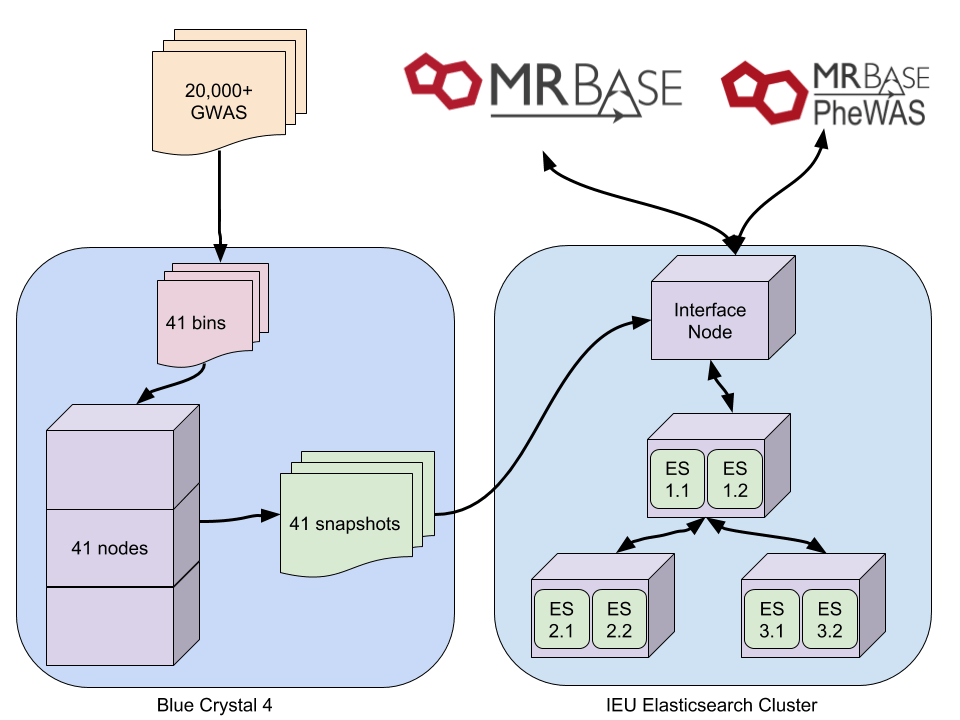
Indexing was being done on our interface node. Up to 30 GWAS were being indexed in parallel but even when initial indexing speed was ok, this was still going to take weeks. We are fortunate here to have some dedicated nodes on a compute cluster, so after much hesitation, we set up the indexing to be done on there. This method involved splitting 20,000 UKBiobank GWAS into groups of 500. Each of these was then indexed independently on a compute node, and after completion a snapshot of each was created. This dropped indexing time from weeks to a single day.
Problem 4
After copying the snapshots to the Elasticsearch cluster, it was hoped that magically they would restore without any of the previous issues. This was not the case. We were still limited by the heap on each instance. Having held off trying to set up multiple Elasticsearch instances on a single node we decided to give it a go. After many hours of hacking at services and config files (a separate blog post maybe) we doubled our number of workers from 3 to 6. This doubled our heap and removed all the issues we were having. Snapshots were restored twice as fast as before, and all 41 were restored in one day.
So, the final process looks like this:
What now?
Currently the main users of this data are via MR-Base (http://www.mrbase.org/), however, the APIs used to query the data are freely available with no quotas (at the moment) and available via TwoSampleMR (https://github.com/MRCIEU/TwoSampleMR). We now have over 250 billion individual genetic associations indexed and ready to search. Our plans are to continue adding data doing our best to ensure fast and reliable queries. Numbers of users and queries are increasing, with over 500 users and 1.5 million queries in the last week. We are also looking at other data options, as certain queries are not optimal using this system. For example, the PheWas browser (http://phewas.mrbase.org/) is searching across all studies, which might be better achieved using BCF files and bcftools. This hybrid system will help remove some of the load off Elasticsearch and enable us to perform global queries that return large amounts of data. In addition, we are testing our platform on Oracle Cloud to test different layouts and configurations.